What is Top-Down Proteomics?
Top-down Proteomics is the analysis of intact proteins by mass spectrometry. This technique allows for exact mass calculations of protein analytes and is thus a reliable method for characterizing proteoforms. Specialized top-down techniques allow for analysis of folded proteins and their non-covalent binding partners, which provides a means for studying endogenous protein complexes.

ApoA-1 acylation in heart disease
ApoA-1 is involved in reverse cholesterol transport. Acylation of ApoA-1 may be associated with decreased risk of heart disease due to changes in membrane affinity. Shown are spectra of intact ApoA-1 bearing an oleoyl lipidation. Fragmentation techniques can be used to localize this modification to a single amino acid residue.
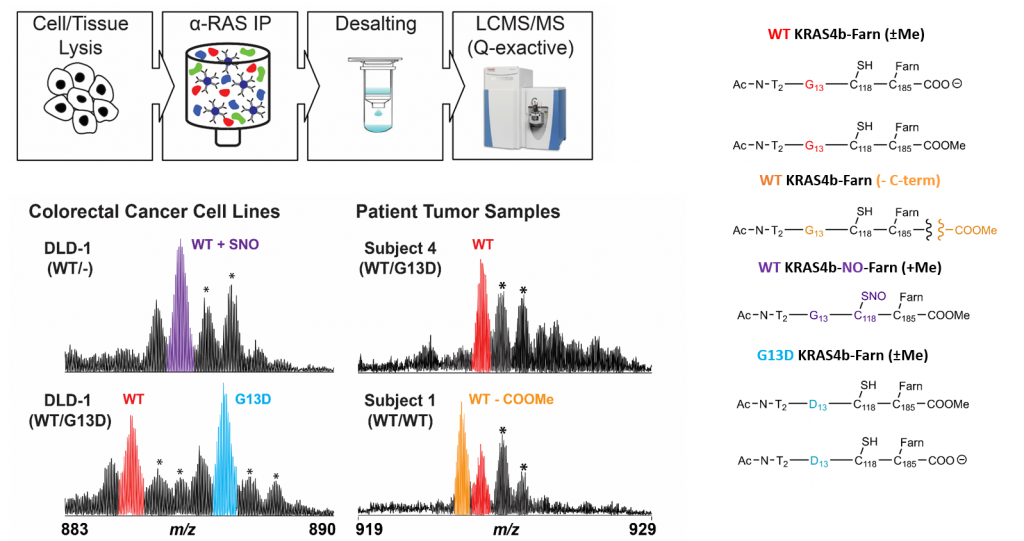
KRAS proteoforms in colon cancer
Mutations of KRAS are associated with numerous cancers; PTMs may further drive pathogenicity. A targeted IP-MS assay can be used to identify KRAS4b proteoforms in colorectal cancer cell line and tumor samples. Shown are spectra of identified modifications. (Proc. Natl. Acad. Sci. 2018, accepted)
What is a Proteoform?
A proteoform is the specific molecular form of a gene product, including any variation due to genetic mutation, alternative RNA splicing, and post-translational modifications (PTMs). Proteoforms have distinct biological functions, and are often difficult to characterize due to their subtle chemical differences. The Kelleher lab emphasizes innovation in proteomics tools and proteoform discovery.